
Final Diagnosis - Amyloid and B-Cell Lymphoma
DIAGNOSIS
MASS, LEFT ORBITAL, EXCISION:
- A. MARKED AMYLOID DEPOSITION.
B. PLASMACYTIC NEOPLASM FAVOR B-CELL LYMPHOMA WITH PLASMACYTIC DIFFERENTIATION.
Diagnostic comment
Morphologic examination demonstrates extensive deposition of amorphous congophilic material consistent with amyloid. There are also scattered foci of lymphocytes with occasional follicles with germinal centers. Associated with these follicles are a moderate number of kappa restricted, CD19+, cyclin D1- and CD56- plasma cells. The possibility of a B-cell lymphoma with plasmacytic differentiation is favored over a plasma cell neoplasm. Correlation with the other clinical, laboratory, and radiology findings, as well as the consideration for a bone marrow biopsy would be of great interest in further characterization of this neoplasm. If additional material is collected, fresh tissue for flow cytometric immunophenotypic studies will be helpful. If clinically indicated, material could be sent out for further characterization of amyloid.
Patient Follow-up
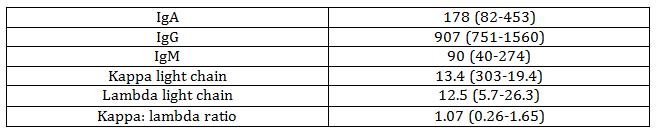
The patient had an additional biopsy sent out for further characterization of amyloid, which showed AL kappa type amyloidosis. Serum immunological studies were performed, with the results shown in Table 2. The patient also had serum and urine immunoelectrophoresis performed, which detected no serum or urine monoclonal proteins, and a lumbar puncture performed, with negative cytology.
Table 2. Immunologic Lab Values
As treatment, the patient received radiation, with a total dose of 24 Gy in 12 fractions using a 3D conformal treatment to cover the entire orbital region. At her follow up after radiotherapy, she noted improved visual acuity, particularly in the left lateral visual field. Repeat CT sinus/orbit showed stable to minimal decrease in size of soft tissue mass, see Figure 7.
The patient also agreed to start 4 courses of systemic chemotherapy with Rituxan.
Due to the amyloidosis being of the AL kappa type, an echocardiogram was performed, which showed no evidence of involvement by amyloidosis.
DISCUSSION
Amyloidosis is the disease caused by accumulation of abnormal extracellular aggregates of low-molecular weight proteins, or amyloid fibrils, in tissues which causes damage of affected organs [1-3]. Light chain amyloidosis (AL) is the most common type of amyloidosis affecting approximately 10 patients per million per year, and is often the most lethal, as it can affect the heart, causing a restrictive cardiomyopathy [1-6]. It is caused by deposition of the immunoglobulin light chain in the setting of plasma cell neoplasms [1-6]. Serum amyloid A, an acute phase reactant, is implicated in Amyloid A amyloidosis (AA), which is associated with chronic inflammatory diseases [1-3, 7-8]. Other types of amyloidosis include hereditary, dialysis-related, systemic old-age, and organ-specific amyloidosis [1-3, 9-12].
The patient's amyloid protein was classified as amyloid light chain, kappa type. The periocular soft tissue mass may represent localized AL amyloidosis, or it is possible that it is the first manifestation of systemic AL amyloidosis, which can be primary, or secondary to plasma cell myeloma, or rarely Waldenstrom macroglobulinemia or non-Hodgkin lymphoma [1-6, 20]. The patient's serum and urine protein electrophoreses were unremarkable, along with the patient's serum immunoglobulin and light chains; absence of serum or urine monoclonal proteins occurs in less than 5% of patients with AL amyloidosis [1]. The clinical manifestations of AL amyloid are varied and depend on which organ is affected. There is commonly renal, cardiac, gastrointestinal, and neurological involvement [1-6].
Diagnosis of amyloidosis usually occurs at biopsy, but imaging can sometimes be suspicious of amyloid deposition [1]. For patients with systemic involvement, fat pad biopsy is the recommended site with sensitivity of 57-85% and a specificity of 92-100% for AL [1, 13, 17-18]. Amyloid deposits that are stained with Congo Red show characteristic apple-green birefringence under polarized light [1-6]. Immunohistochemistry can be useful in characterizing amyloid type, particularly for amyloid A and transthyretin amyloid, but has less utility with AL amyloid, as the antigenic epitopes may be lost due to proteolysis during deposition and fibril formation [1, 19]. Additional methods for characterizing amyloid type include mass spectroscopy and amino acid sequencing, which can be performed on formalin-fixed paraffin-embedded tissue via laser capture microdissection [1, 15].
Treatment of AL amyloidosis usually involves treating the underlying plasma cell neoplasm [1-6]. Options for treatment include chemotherapeutics, autologous stem cell transplantation, and radiotherapy for local involvement, among others [1-6, 20]. Our patient received local radiotherapy combined with Rituxin. Treatment with Rituxin, particularly in combination with other agents such as idelalisib has shown efficacy in systemic AL [21].
REFERENCES
- Gorevic PD, Shur PH, and Roman PL. Overview of amyloidosis. UpToDate.
- Chiti F, Dobson CM. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem 2017; 86:27.
- Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221.
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 2011; 97:75.
- Merlini G , Wechalekar AD and Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013 121:5124-5130; doi: https://doi.org/10.1182/blood-2013-01-453001
- Merlini G, Comenzo RL, Seldin DC, et al. Immunoglobulin light chain amyloidosis. Expert Rev Hematol 2014; 7:143.
- Pinney JH, Lachmann HJ. Systemic AA amyloidosis. Subcell Biochem 2012; 65:541.
- Obici L, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmun Rev 2012; 12:14.
- Pinney JH, Whelan CJ, Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013; 2:e000098.
- Miura Y, Harumiya S, Ono K, et al. Galectin-7 and actin are components of amyloid deposit of localized cutaneous amyloidosis. Exp Dermatol 2013; 22:36.
- Said SM, Sethi S, Valeri AM, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol 2013; 8:1515.
- Hamidi Asl K, Liepnieks JJ, Nakamura M, Benson MD. Organ-specific (localized) synthesis of Ig light chain amyloid. J Immunol 1999; 162:5556.
- Westermark P. Subcutaneous adipose tissue biopsy for amyloid protein studies. Methods Mol Biol 2012; 849:363.
- Arbustini E, Morbini P, Verga L, Merlini G. Light and electron microscopy immunohistochemical characterization of amyloid deposits. Amyloid 1997; 4:157.
- Vrana JA, Gamez JD, Madden BJ, et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 2009; 114:4957.
- Arbustini E, Morbini P, Verga L, Merlini G. Light and electron microscopy immunohistochemical characterization of amyloid deposits. Amyloid 1997; 4:157.
- Duston MA, Skinner M, Meenan RF, Cohen AS. Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum 1989; 32:82.
- van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 2006; 54:2015.
- Davern S, Tang LX, Williams TK, et al. Immunodiagnostic capabilities of anti-free immunoglobulin light chain monoclonal antibodies. Am J Clin Pathol 2008; 130:702.
- Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica. 2014 Feb; 99(2): 209-221.
- Visentin A et al. Idelalisib plus rituximab is effective in systemic AL amyloidosis secondary to chronic lymphocytic leukaemia. Hematol Oncol. 2018 Feb;36(1):366-369. doi: 10.1002/hon.2480. Epub 2017 Oct 3.
Contributed by Terri Jones, MD and Nidhi Aggarwal, MD